We have employed explicit molecular dynamics (MD) simulations as well as coarse-grained molecular approaches, including electrostatic potential (EP) calculations, docking, etc. to study the behavior of protein retention on ion-exchange and multimodal (MM) systems.m
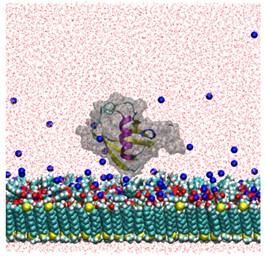
The goal of these studies is to provide fundamental understanding of the nature of water-mediated protein-ligand interactions at a molecular level which can provide significant insight into the design of MM ligands, the roles of synergy and the modulation of selectivity using fluid phase modifiers (FPMs). This can further be used to explain, and indeed predict, the retention behavior of proteins on multimodal resins under a variety of solution conditions1-6.
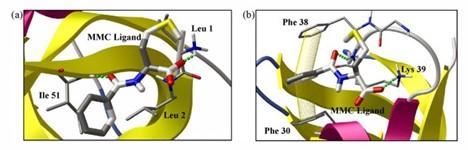
Molecular docking has been employed to examine the various interactions between the protein CspB and ligands in free solution, and to study the modes of interaction between MM ligands and ubiquitin¹.
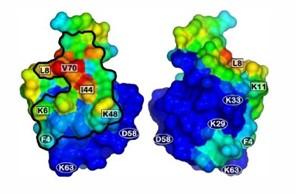
We have employed all-atom MD simulations of ubiquitin with multimodal ligands in free solution to identify binding preferences and to shed light on the “pseudo-affinity” nature of multimodal interactions. The results showed a preferred binding face on the protein, which corroborated the results obtained from NMR and chromatography experiments² .
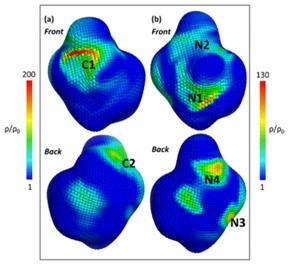
Atomistic molecular dynamics (MD) simulations were employed to study the interaction of two multimodal chromatographic ligands with proteins. Spherical harmonic expansion was used to analyze ligand density of Capto MMC and Nuvia CPrime ligand which contain similar chemical moieties—aromatic, carboxyl, and amide—but have different structural arrangements. We have found that the Capto MMC ligand, which contains an additional aliphatic group, and has greater structural flexibility displays stronger interactions than Nuvia CPrime ligand. These differences in binding affinities and modalities for multimodal ligands can result in significantly different binding behavior towards proteins with important implications for bioprocessing³.
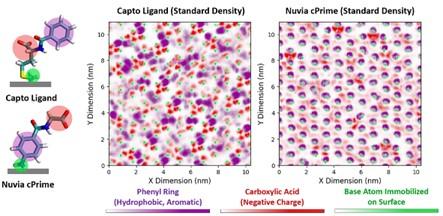
Molecular dynamics simulations have revealed that Capto MMC ligands that are flexible and terminate in a hydrophobic group tend to cluster on the surface, while less flexible Nuvia CPrime ligands without a terminal hydrophobic group do not cluster. This results in the formation of a surface pattern for Capto MMC surfaces containing large patches of hydrophobicity and charge whose sizes exceeded the length scale of the individual ligands⁴⁻⁵.
(Cr: Dr. Imee Sinha; Dr. Mayank Vats)
Selected References:
1.Chung, Wai Keen, et al. "Evaluation of protein adsorption and preferred binding regions in multimodal chromatography using NMR." Proceedings of the National Academy of Sciences 107.39 (2010): 16811-16816.
2.Freed, Alexander S., Shekhar Garde, and Steven M. Cramer. "Molecular simulations of multimodal ligand–protein binding: elucidation of binding sites and correlation with experiments." The Journal of Physical Chemistry B 115.45 (2011): 13320-13327.
3.Parimal, Siddharth, Shekhar Garde, and Steven M. Cramer. "Interactions of multimodal ligands with proteins: insights into selectivity using molecular dynamics simulations." Langmuir 31.27 (2015): 7512-7523.
4.Bilodeau, Camille L., et al. "Conformational equilibria of multimodal chromatography ligands in water and bound to protein surfaces." The Journal of Physical Chemistry B 123.23 (2019): 4833-4843.
5.Bilodeau, Camille L., et al. "Formation of ligand clusters on multimodal chromatographic surfaces." Langmuir 35.51 (2019): 16770-16779.
6.Vats, Mayank. Elucidating the role of multimodal ligand surfaces in protein chromatography using molecular dynamics simulations. Diss. Rensselaer Polytechnic Institute, Troy, NY, 2022.