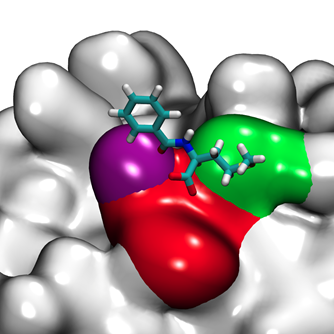
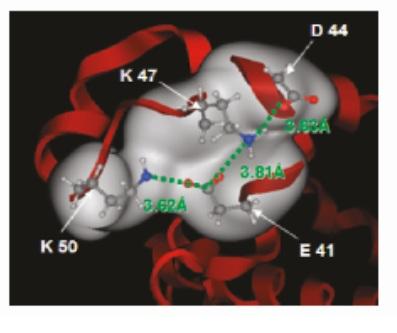
Molecular simulations, coursed grained modeling, mechanistic and hybrid models and fully data driven modeling are used to provide insight into the interactions in these complex bioseparation systems and to develop in silico prediction tools for process development and the discovery of new separation materials.